Everyone knows that the use of Large Language Models is in its infancy. Successes are often stunning as the LLM returns answers that are better than expected. Failures happen, too. They can be frustrating.
The following sequence of prompts and responses show how the LLM (Claude) eventually fixed a failing response. In this case, all that was needed was to supply the LLM with the error message generated by the failure. Each time a new version of R code was delivered. After three tries, the modifications worked.
The success prompted some follow-on queries that allow further testing of the R code.
The test bed is a form of analysis used in Vegetation Ecology. The Point-Centered Quadrat method is a niche tool. It seemed like a likely candidate for testing.
Note that no artifacts are used in this sequence of runs.
Point-Centered Quadrat Method
Prompt
Can you explain the point-centered quadrat method used in vegetation analysis?
Response
The point-centered quadrat method is a sampling technique used in vegetation analysis to assess plant distribution and density. Here’s a concise explanation:
Sampling points are randomly or systematically placed across the study area.
At each point, imaginary lines are drawn to divide the area into four quadrants.
In each quadrant, the distance from the center point to the nearest plant is measured.
The species of each measured plant is recorded.
This process is repeated for all sampling points.
The data collected allows researchers to calculate plant density, frequency, and relative abundance.
This method is efficient for surveying large areas and is particularly useful for sparse vegetation where traditional quadrat methods might be less effective.
Would you like me to elaborate on any specific aspect of this method, such as its advantages, limitations, or calculation techniques?
Prompt
Can you create some R code to show how to analyze data from this methodology?
Response
Certainly, I can create some R code to demonstrate how to analyze data from the point-centered quadrat method. I’ll create an artifact with a sample R script that includes data import, basic analysis, and visualization.
This R script demonstrates how to analyze data from the point-centered quadrat method. Here’s a brief explanation of what the code does:
It starts by creating a sample dataset that mimics data collected using the point-centered quadrat method.
It calculates key metrics such as density, relative density, frequency, and importance value for each species.
It visualizes the results using a bar plot of importance values.
It calculates and reports diversity indices (Shannon diversity and evenness).
Finally, it performs an ordination analysis using Non-metric Multidimensional Scaling (NMDS) to visualize community composition.
To use this code with your own data, you would replace the sample data creation step with importing your actual data. The rest of the analysis can then be applied to your dataset.
Would you like me to explain any specific part of this analysis in more detail?
Code Failures
The R code didn’t work. The following prompts show the error message given at each failure. The code and responses are abbreviated here. Basically, prompting with an error message yielded a new chunk of R code. When that was run, if there was an error, the error message was used in the next prompt. This was done until the code worked.
In retrospect, it’s pretty clear that the working code was not used in a subsequent run; Claude seemed to revert to a previous version. This simply emphasizes that we’re early in the development of this tool and we need to be alert to unexpected results.
Prompt (after 1st failure)
I’m getting the following error message: “““Error in UseMethod(”isSymmetric”) : no applicable method for ‘isSymmetric’ applied to an object of class “table”““”
Prompt (after 2nd failure)
I still get the same message. Any new ideas?
Prompt (after 3rd failure)
It’s better. But now I get this message (after the function shown):“““nmds_scores <- as.data.frame(scores(nmds_result)) Error in (function (…, row.names = NULL, check.rows = FALSE, check.names = TRUE, : arguments imply differing number of rows: 50, 4”“”
Response (it works!)
Show the code
# Load required librarieslibrary(tidyverse)library(vegan)# Create sample dataset.seed(123)pcq_data<-data.frame( point =rep(1:50, each =4), quadrant =rep(1:4, 50), species =sample(c("Species A", "Species B", "Species C", "Species D"), 200, replace =TRUE), distance =runif(200, 0, 10))# Calculate densitytotal_points<-length(unique(pcq_data$point))mean_distance<-mean(pcq_data$distance)area<-(mean_distance^2)*4*total_pointsdensity<-nrow(pcq_data)/area# Calculate relative density for each speciesspecies_counts<-table(pcq_data$species)relative_density<-species_counts/sum(species_counts)*100# Calculate frequencyspecies_frequency<-sapply(unique(pcq_data$species), function(sp){sum(tapply(pcq_data$species==sp, pcq_data$point, any))/total_points})# Calculate importance valueimportance_value<-relative_density+(species_frequency/sum(species_frequency)*100)# Combine resultsresults<-data.frame( species =names(species_counts), count =as.vector(species_counts), relative_density =as.vector(relative_density), frequency =as.vector(species_frequency), importance_value =as.vector(importance_value))# Print resultsprint(results)
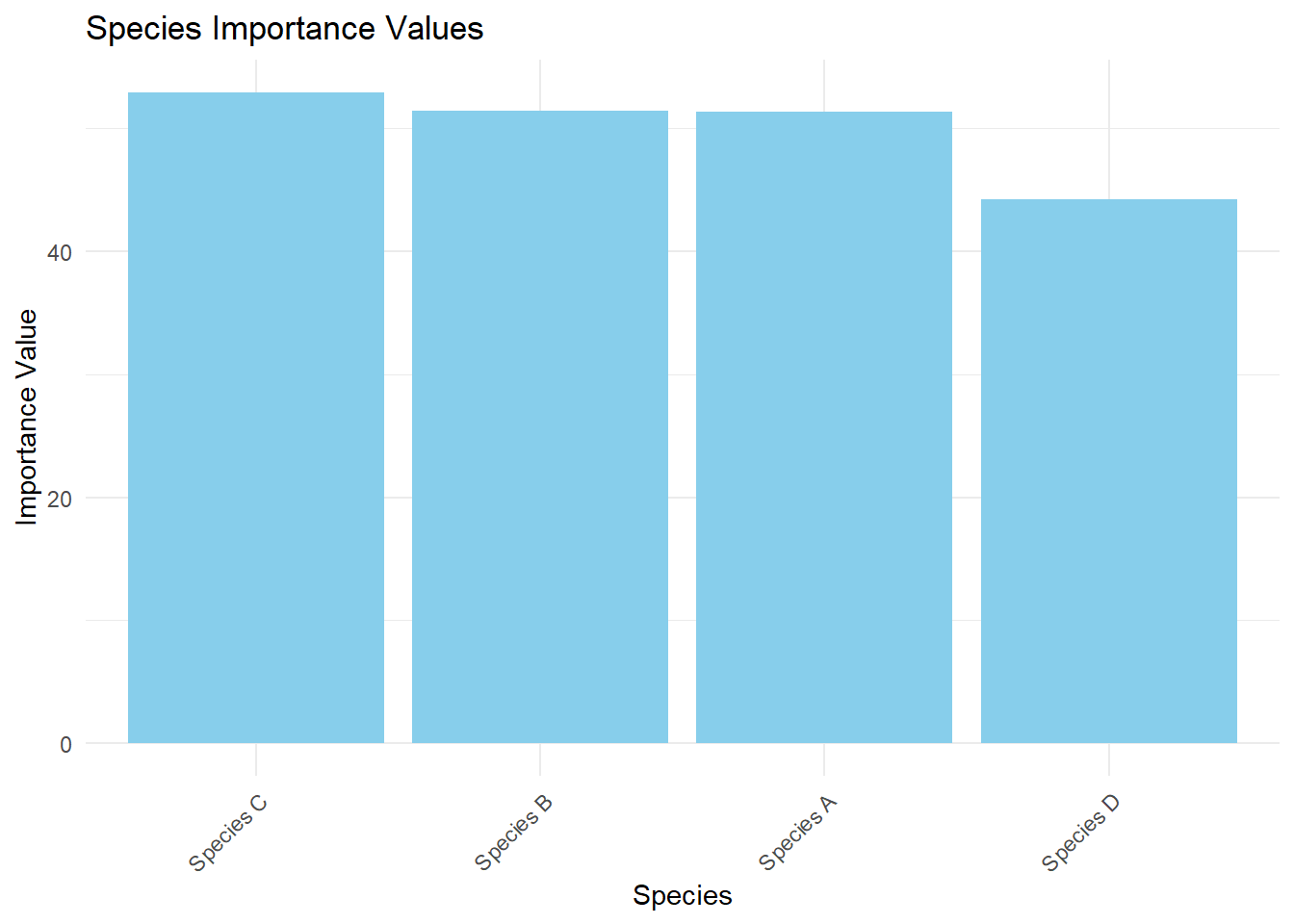
species count relative_density frequency importance_value
1 Species A 45 22.5 0.82 51.37324
2 Species B 55 27.5 0.68 51.44366
3 Species C 58 29.0 0.68 52.94366
4 Species D 42 21.0 0.66 44.23944
Show the code
# Visualize resultsggplot(results, aes(x =reorder(species, -importance_value), y =importance_value))+geom_bar(stat ="identity", fill ="skyblue")+labs(title ="Species Importance Values", x ="Species", y ="Importance Value")+theme_minimal()+theme(axis.text.x =element_text(angle =45, hjust =1))
Show the code
# Calculate and plot species diversitydiversity_indices<-diversity(species_counts, index ="shannon")evenness<-diversity_indices/log(length(species_counts))cat("Shannon diversity index:", diversity_indices, "\n")
Run 0 stress 0.05058811
Run 1 stress 0.04343727
... New best solution
... Procrustes: rmse 0.05678532 max resid 0.2076734
Run 2 stress 0.0941133
Run 3 stress 0.0524645
Run 4 stress 0.05246442
Run 5 stress 0.09046666
Run 6 stress 0.04315295
... New best solution
... Procrustes: rmse 0.01782708 max resid 0.09147472
Run 7 stress 0.08968852
Run 8 stress 0.1012873
Run 9 stress 0.05257916
Run 10 stress 0.04315191
... New best solution
... Procrustes: rmse 0.002784508 max resid 0.01853647
Run 11 stress 0.04315204
... Procrustes: rmse 0.001310195 max resid 0.008663865
... Similar to previous best
Run 12 stress 0.05058801
Run 13 stress 0.05257738
Run 14 stress 0.08968846
Run 15 stress 0.08968867
Run 16 stress 0.1017777
Run 17 stress 0.05248184
Run 18 stress 0.05058808
Run 19 stress 0.04315179
... New best solution
... Procrustes: rmse 0.000315043 max resid 0.001927479
... Similar to previous best
Run 20 stress 0.0525793
*** Best solution repeated 1 times
Show the code
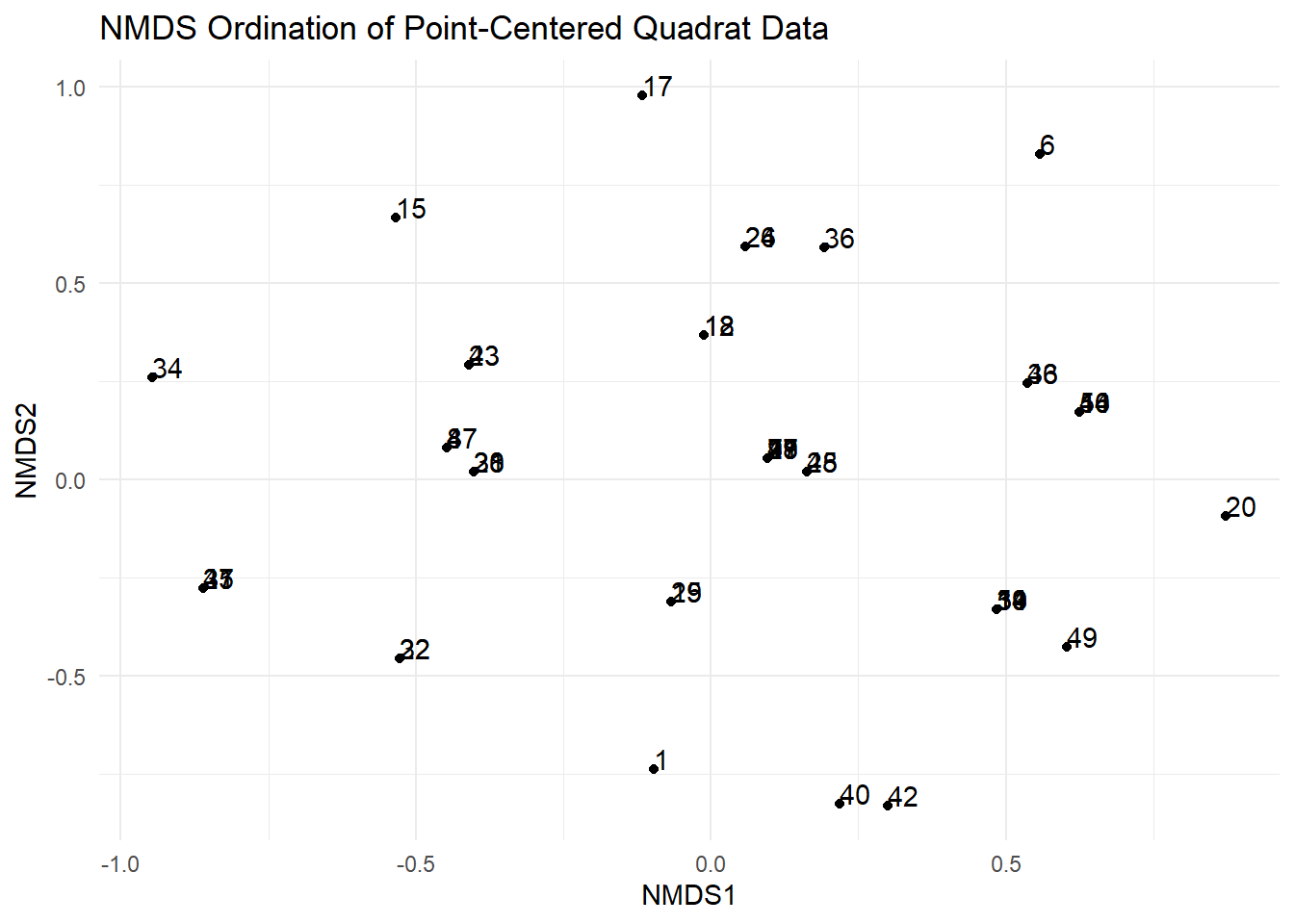
# Extract NMDS scores correctlynmds_scores<-as.data.frame(scores(nmds_result, display ="sites"))nmds_scores$point<-rownames(nmds_scores)# Create a more informative NMDS plot using ggplot2ggplot(nmds_scores, aes(x =NMDS1, y =NMDS2, label =point))+geom_point()+geom_text(hjust =0, vjust =0)+labs(title ="NMDS Ordination of Point-Centered Quadrat Data", x ="NMDS1", y ="NMDS2")+theme_minimal()
At this point, with a working system, there were several question regarding how to supply data for real analyses and some guidance on how to interpret the results.
Prompt
Can you print part of the input data in a table format so I can see better how to enter my data?
Response
Certainly! I’ll modify the code to print a sample of the input data in a table format. This will help you understand how to structure your own data for analysis.
Point-Centered Quadrat Analysis in R
I’ve added code to print the first 20 rows of the input data using the kable function from the knitr package, which produces a nicely formatted table. This will give you a clear view of how the data should be structured.
Here’s an explanation of the data structure:
point: This represents the sampling point number. In a real study, this would be a unique identifier for each sampling location.
quadrant: This represents the quadrant number (1-4) within each sampling point.
species: This is the species name or identifier found in each quadrant.
distance: This is the distance from the center point to the nearest plant of the specified species in that quadrant.
To use this script with your own data, you should structure your data in a similar format:
Each row represents one measurement (one plant in one quadrant of one sampling point).
You should have four rows for each sampling point (one for each quadrant).
Make sure your species names are consistent throughout the dataset.
Distances should be numeric values.
You can either create this data frame directly in R (if you have a small dataset) or, more commonly, you would import it from a CSV file using a command like:
Show the code
# Load required librarieslibrary(tidyverse)library(vegan)library(knitr)# For nice table formatting# Create sample dataset.seed(123)pcq_data<-data.frame( point =rep(1:50, each =4), quadrant =rep(1:4, 50), species =sample(c("Species A", "Species B", "Species C", "Species D"), 200, replace =TRUE), distance =round(runif(200, 0, 10), 2))# Print the first 20 rows of the input datacat("Sample of input data:\n")
| point| quadrant|species | distance|
|-----:|--------:|:---------|--------:|
| 1| 1|Species C | 2.39|
| 1| 2|Species C | 9.62|
| 1| 3|Species C | 6.01|
| 1| 4|Species B | 5.15|
| 2| 1|Species C | 4.03|
| 2| 2|Species B | 8.80|
| 2| 3|Species B | 3.64|
| 2| 4|Species B | 2.88|
| 3| 1|Species C | 1.71|
| 3| 2|Species A | 1.72|
| 3| 3|Species D | 4.82|
| 3| 4|Species B | 2.53|
| 4| 1|Species B | 2.16|
| 4| 2|Species A | 6.74|
| 4| 3|Species B | 0.48|
| 4| 4|Species C | 7.01|
| 5| 1|Species D | 3.52|
| 5| 2|Species A | 4.09|
| 5| 3|Species C | 8.21|
| 5| 4|Species C | 9.19|
Show the code
# Rest of the analysis code remains the same# ...# Calculate densitytotal_points<-length(unique(pcq_data$point))mean_distance<-mean(pcq_data$distance)area<-(mean_distance^2)*4*total_pointsdensity<-nrow(pcq_data)/area# Calculate relative density for each speciesspecies_counts<-table(pcq_data$species)relative_density<-species_counts/sum(species_counts)*100# Calculate frequencyspecies_frequency<-sapply(unique(pcq_data$species), function(sp){sum(tapply(pcq_data$species==sp, pcq_data$point, any))/total_points})# Calculate importance valueimportance_value<-relative_density+(species_frequency/sum(species_frequency)*100)# Combine resultsresults<-data.frame( species =names(species_counts), count =as.vector(species_counts), relative_density =as.vector(relative_density), frequency =as.vector(species_frequency), importance_value =as.vector(importance_value))# Print resultsprint(results)
species count relative_density frequency importance_value
1 Species A 45 22.5 0.82 51.37324
2 Species B 55 27.5 0.68 51.44366
3 Species C 58 29.0 0.68 52.94366
4 Species D 42 21.0 0.66 44.23944
Show the code
# ... (rest of the code remains unchanged)
Prompt
Can you make a dataset that has several quadrats with “atypical” data so I can see how the looks in the analysis results?
Response
Certainly! I’ll create a dataset that includes some “atypical” data to show how it affects the analysis results. We’ll include scenarios like missing species in some quadrats, extremely distant plants, and highly clustered species.
Point-Centered Quadrat Analysis with Atypical Data
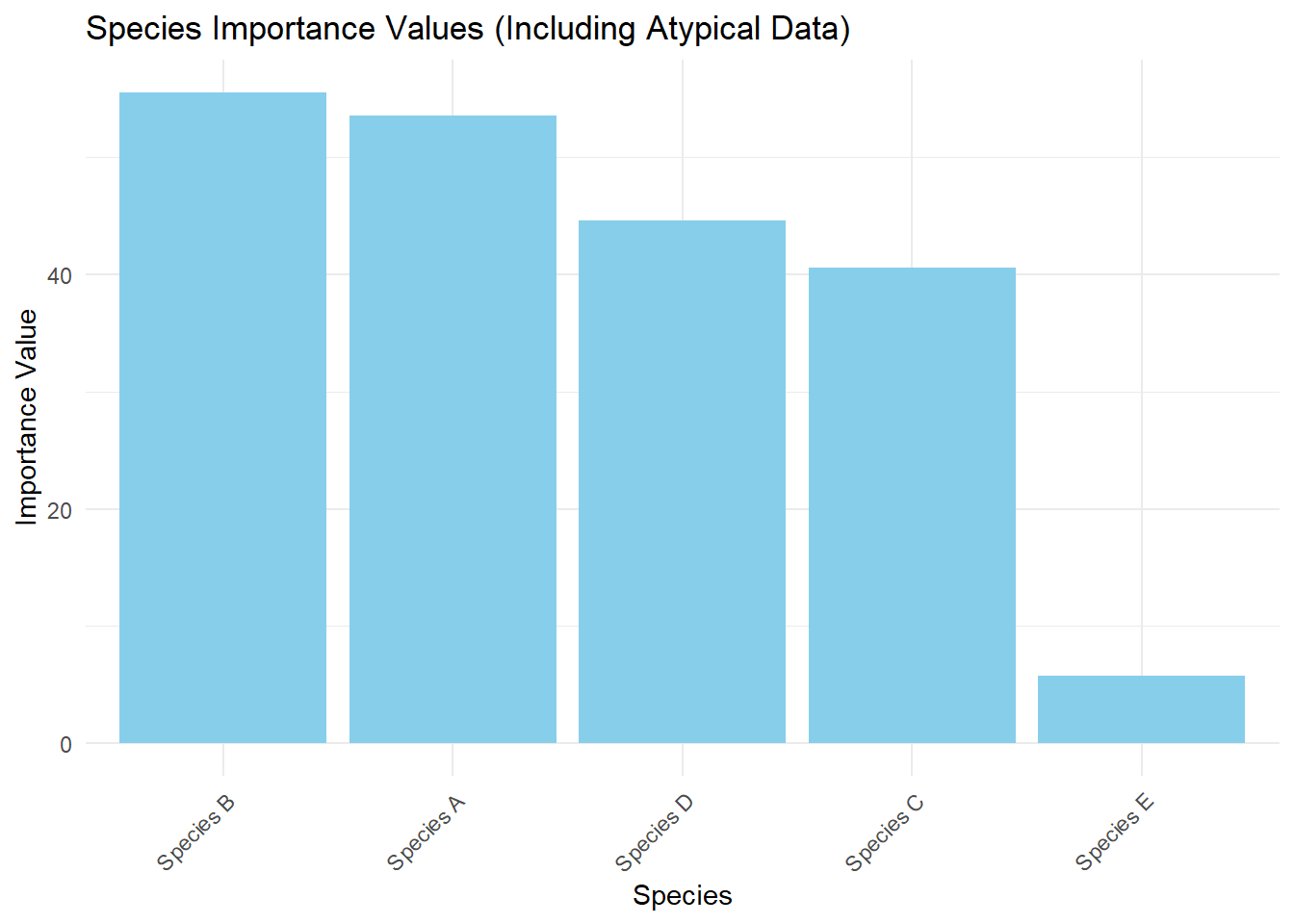
This script creates a dataset with some atypical patterns:
Points 1-40 have normal, randomly distributed data.
Points 41-42 have a clustered rare species (Species E) at very close distances.
Point 43 has empty quadrats (NA values).
Points 44-46 have a highly dominant species (Species A) at large distances.
Points 47-50 have a moderately dominant species (Species B) at medium distances.
Here’s how these atypical patterns might affect your analysis:
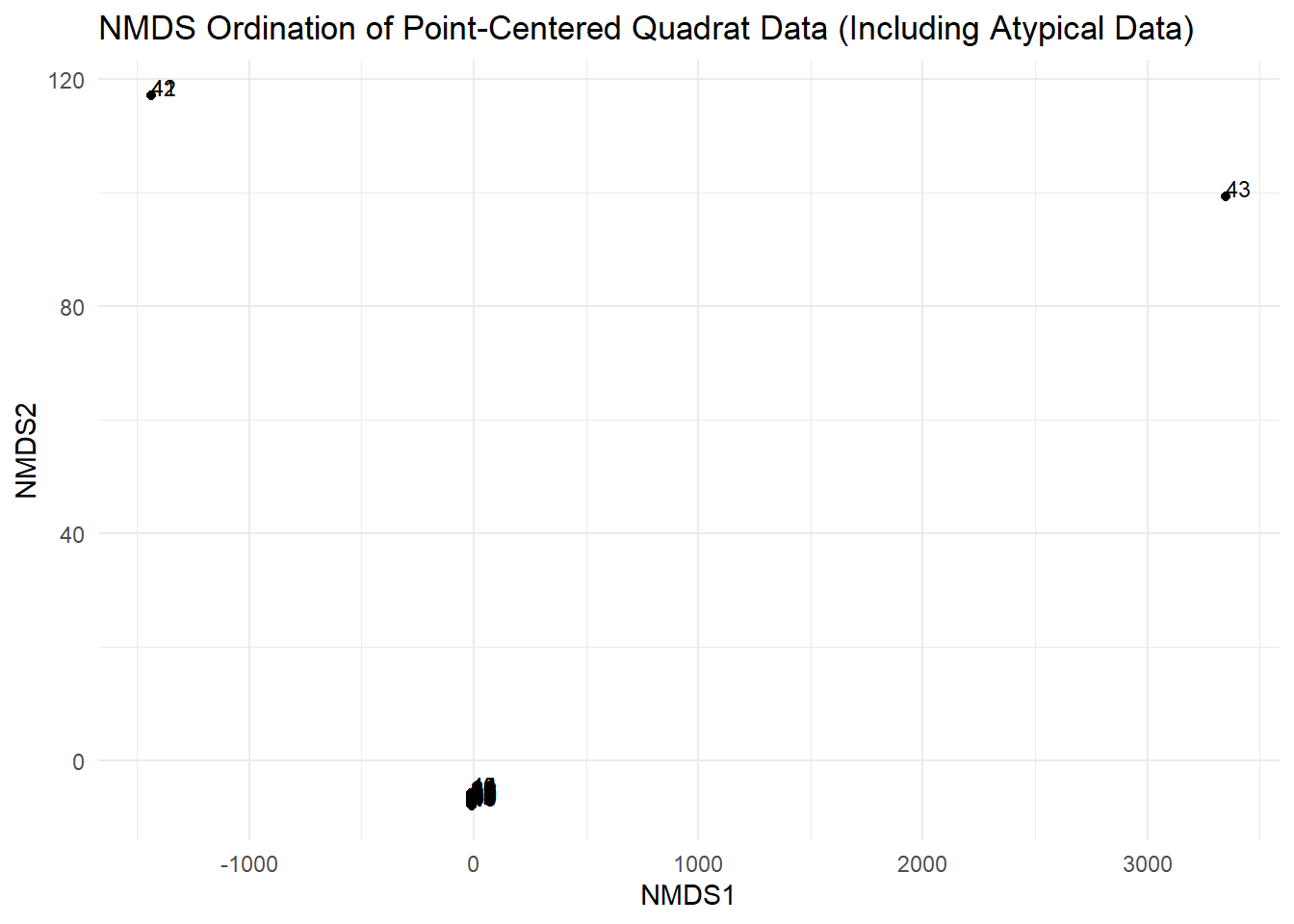
Rare, clustered species (Species E):
- Will have low relative density and frequency.
- May appear as an outlier in the NMDS plot.
Empty quadrats:
- Will slightly reduce overall density estimates.
- May affect diversity calculations.
Highly dominant species at large distances (Species A):
- Will have high relative density and frequency.
- May skew the importance value calculations.
Moderately dominant species (Species B):
- Will have moderately high relative density and frequency.
When you run this script, pay attention to:
The “Analysis Results” table: Look at how Species E (rare), Species A (dominant), and Species B (moderately dominant) compare in terms of count, relative density, frequency, and importance value.
The Importance Values plot: Notice how the atypical species are represented.
The NMDS plot: Look for clustering or outliers, especially for points 41-50 which contain the atypical data.
The diversity indices: These may be affected by the presence of rare and dominant species.
The NMDS stress value: This might be higher due to the atypical data patterns.
This example demonstrates how the point-centered quadrat method handles various data anomalies. In real-world scenarios, such patterns might indicate interesting ecological phenomena or sampling issues that warrant further investigation.
Would you like me to explain any specific part of the results or how these atypical patterns are reflected in the analysis?
Show the code
# Load required librarieslibrary(tidyverse)library(vegan)library(knitr)# Create sample data with atypical patternsset.seed(456)normal_data<-data.frame( point =rep(1:40, each =4), quadrant =rep(1:4, 40), species =sample(c("Species A", "Species B", "Species C", "Species D"), 160, replace =TRUE), distance =round(runif(160, 0, 10), 2))# Add atypical dataatypical_data<-data.frame( point =rep(41:50, each =4), quadrant =rep(1:4, 10), species =c(rep("Species E", 8), # Clustered rare speciesrep(NA, 4), # Empty quadratsrep("Species A", 12), # Highly dominant speciesrep("Species B", 16)# Moderately dominant species), distance =c(runif(8, 0, 1), # Clustered species (very close)rep(NA, 4), # Empty quadratsrunif(12, 8, 10), # Distant dominant speciesrunif(16, 3, 7)# Moderately distant species))pcq_data<-rbind(normal_data, atypical_data)# Print the last 30 rows of the input data to show atypical patternscat("Sample of input data (showing atypical patterns):\n")
# Create species matrix for NMDSspecies_matrix<-pcq_data%>%group_by(point, species)%>%summarise(count =n(), .groups ="drop")%>%pivot_wider(names_from =species, values_from =count, values_fill =0)%>%column_to_rownames("point")# Remove the NA column if presentspecies_matrix<-species_matrix[, !is.na(colnames(species_matrix))]# Perform ordination analysis (NMDS)nmds_result<-metaMDS(species_matrix, trymax =100)
Run 0 stress 0.008750079
Run 1 stress 0.002520559
... New best solution
... Procrustes: rmse 0.0919235 max resid 0.3069367
Run 2 stress 0.001526668
... New best solution
... Procrustes: rmse 0.1086397 max resid 0.6368838
Run 3 stress 0.001138746
... New best solution
... Procrustes: rmse 0.04080056 max resid 0.1971615
Run 4 stress 0.002334596
Run 5 stress 0.0004422428
... New best solution
... Procrustes: rmse 0.08678562 max resid 0.3958227
Run 6 stress 0.0009746293
Run 7 stress 0.001233199
Run 8 stress 0.0009002158
... Procrustes: rmse 0.07480737 max resid 0.3624818
Run 9 stress 9.890448e-05
... New best solution
... Procrustes: rmse 0.06876043 max resid 0.4453528
Run 10 stress 0.001288601
Run 11 stress 0.001830215
Run 12 stress 9.908735e-05
... Procrustes: rmse 0.1171855 max resid 0.4970956
Run 13 stress 0.004376256
Run 14 stress 0.004066578
Run 15 stress 0.004271741
Run 16 stress 9.778766e-05
... New best solution
... Procrustes: rmse 0.08626762 max resid 0.3708633
Run 17 stress 0.0001956243
... Procrustes: rmse 0.03081051 max resid 0.1377626
Run 18 stress 9.71747e-05
... New best solution
... Procrustes: rmse 0.04487283 max resid 0.234499
Run 19 stress 0.003288032
Run 20 stress 0.0007277387
Run 21 stress 0.004673706
Run 22 stress 0.0007831094
Run 23 stress 9.589267e-05
... New best solution
... Procrustes: rmse 0.05780483 max resid 0.3315277
Run 24 stress 9.749894e-05
... Procrustes: rmse 0.05028861 max resid 0.2586076
Run 25 stress 0.004667348
Run 26 stress 0.002558886
Run 27 stress 0.001491336
Run 28 stress 0.003274536
Run 29 stress 0.004056193
Run 30 stress 9.848379e-05
... Procrustes: rmse 0.06670758 max resid 0.3382634
Run 31 stress 0.001934921
Run 32 stress 0.001172622
Run 33 stress 0.003689473
Run 34 stress 0.002920627
Run 35 stress 0.00434644
Run 36 stress 0.0001090552
... Procrustes: rmse 0.007611328 max resid 0.0421087
Run 37 stress 0.003498463
Run 38 stress 0.003872278
Run 39 stress 0.002663436
Run 40 stress 0.0004325499
... Procrustes: rmse 0.03452009 max resid 0.2176675
Run 41 stress 0.003398822
Run 42 stress 0.006415283
Run 43 stress 0.0006890925
Run 44 stress 0.001821138
Run 45 stress 0.004998745
Run 46 stress 0.002495006
Run 47 stress 0.000899314
Run 48 stress 9.75642e-05
... Procrustes: rmse 0.0241102 max resid 0.1430594
Run 49 stress 0.00221837
Run 50 stress 9.918049e-05
... Procrustes: rmse 0.05765386 max resid 0.3485553
Run 51 stress 0.002052681
Run 52 stress 0.0001420754
... Procrustes: rmse 0.07682426 max resid 0.3930336
Run 53 stress 0.00129011
Run 54 stress 0.0006121333
Run 55 stress 9.484414e-05
... New best solution
... Procrustes: rmse 0.02364272 max resid 0.1307858
Run 56 stress 0.002387283
Run 57 stress 0.001608554
Run 58 stress 0.001623045
Run 59 stress 9.774542e-05
... Procrustes: rmse 0.1277213 max resid 0.5542328
Run 60 stress 0.002582508
Run 61 stress 0.0005647567
... Procrustes: rmse 0.08372655 max resid 0.4681928
Run 62 stress 0.002874892
Run 63 stress 0.0009475414
Run 64 stress 0.002435838
Run 65 stress 0.003617477
Run 66 stress 0.004917061
Run 67 stress 9.57077e-05
... Procrustes: rmse 0.09480989 max resid 0.4586874
Run 68 stress 0.004292835
Run 69 stress 9.881932e-05
... Procrustes: rmse 0.02060286 max resid 0.1009203
Run 70 stress 0.001664599
Run 71 stress 6.514336e-05
... New best solution
... Procrustes: rmse 0.1269227 max resid 0.5274248
Run 72 stress 0.004087057
Run 73 stress 0.001480746
Run 74 stress 0.006102677
Run 75 stress 9.831147e-05
... Procrustes: rmse 0.07249164 max resid 0.3657383
Run 76 stress 9.632722e-05
... Procrustes: rmse 0.06267725 max resid 0.3010556
Run 77 stress 9.829339e-05
... Procrustes: rmse 0.04483209 max resid 0.2332381
Run 78 stress 0.002172876
Run 79 stress 0.002390377
Run 80 stress 0.004723325
Run 81 stress 0.005528413
Run 82 stress 0.003301879
Run 83 stress 0.0002654863
... Procrustes: rmse 0.1070489 max resid 0.4952354
Run 84 stress 0.0001868593
... Procrustes: rmse 0.122976 max resid 0.5611981
Run 85 stress 0.001011643
Run 86 stress 0.006096893
Run 87 stress 0.001205929
Run 88 stress 9.98356e-05
... Procrustes: rmse 0.08097398 max resid 0.3954186
Run 89 stress 0.001955737
Run 90 stress 0.005021377
Run 91 stress 0.002294385
Run 92 stress 0.002334262
Run 93 stress 0.002092417
Run 94 stress 0.001395494
Run 95 stress 0.001396876
Run 96 stress 0.004674485
Run 97 stress 0.0007051011
Run 98 stress 0.001997447
Run 99 stress 0.0005055781
... Procrustes: rmse 0.1008778 max resid 0.4706
Run 100 stress 9.825138e-05
... Procrustes: rmse 0.1212808 max resid 0.6254439
*** Best solution was not repeated -- monoMDS stopping criteria:
79: no. of iterations >= maxit
19: stress < smin
2: scale factor of the gradient < sfgrmin
Show the code
# Extract NMDS scoresnmds_scores<-as.data.frame(scores(nmds_result, display ="sites"))nmds_scores$point<-rownames(nmds_scores)# Create NMDS plotggplot(nmds_scores, aes(x =NMDS1, y =NMDS2, label =point))+geom_point()+geom_text(hjust =0, vjust =0, size =3)+labs(title ="NMDS Ordination of Point-Centered Quadrat Data (Including Atypical Data)", x ="NMDS1", y ="NMDS2")+theme_minimal()